I always forget the concept of q value.
several month ago I found web site with good explanation of q value. but I cannot remember that site, so in this post I record the track for understanding q value.
how to interpret q value
http://www.nonlinear.com/support/progenesis/samespots/faq/pq-values.aspx
how to calculate q value
http://courses.ttu.edu/isqs6348-westfall/images/6348/BonHolmBenHoch.htm
Friday, July 30, 2010
Tuesday, July 27, 2010
velvet VS newbler
To make sure which perform more accurately, I tested two programs in two strategies. Actually the reason why I did do this test is that most of people in my firm say that for de novo assembly FLX is much more suitable than solexa without any evidence. So I decided to test two system, but there is no real data which I can use, so I just do simulation test of the two program which is representative for each system.
Both of them, velvet and newbler are used for de novo assembly. In case of velvet, by using De Bruijn graph methodology It carry out short read assembly with data from solexa, solid. On the other hand, newbler is software from Roche, FLX and it is based on overlap layout consensus methodology (for seeing about the algorithm refer http://www.ncbi.nlm.nih.gov/pubmed/20211242).
I will compare the results from both program in two strategies. All of read data in these tests are simulation data which were made from reference genome by computational simulation.
First, velvet with paired-end read of which length is 78 bp and insert size is 300. newbler with single read of which length is 300 bp.
second, in case of velvet from first test adding long insert library with same condition
Both of them, velvet and newbler are used for de novo assembly. In case of velvet, by using De Bruijn graph methodology It carry out short read assembly with data from solexa, solid. On the other hand, newbler is software from Roche, FLX and it is based on overlap layout consensus methodology (for seeing about the algorithm refer http://www.ncbi.nlm.nih.gov/pubmed/20211242).
I will compare the results from both program in two strategies. All of read data in these tests are simulation data which were made from reference genome by computational simulation.
First, velvet with paired-end read of which length is 78 bp and insert size is 300. newbler with single read of which length is 300 bp.
second, in case of velvet from first test adding long insert library with same condition
Monday, July 26, 2010
stubborness
나이가 먹어가면서 느끼는.. 살아가면서 꼭 필요한 요소가 긍정, 열정, 완고함이라고 생각한다.
긍정의 요소가 있어가 어떠한 일에 대해 가능성을 느낄수 있고 열정의 요소가 있어야 그 가능성을 찾으려는 의지가 생기고 완고함이 있어야 발견한 가능성을 실현 할수 있는것 같다.
꼭 필요하다고 느끼는 이 세가지 모두는 내게 그리 많이 주어지지 않았다. 특히나 이번엔 완고함에 대해 이야기 하고자 한다. 머리속에서 지워지지 않은 일이 있으므로..
완고함이라 함은 어찌보면 의지이기도 하고 믿음이기도 하다. 내가 원하는걸 해낼거라는 의지이자 내가 하는 일이 옳은 일이라고 생각하는 믿음.
어제 일이다. 하루종일 까페에 앉아서 내가 원하는 공부를 하고 내가 원하는 논문을 읽고 나름 보람찬 하루를 보낸 후, 늦은 저녁에 여자 친구와 함께 여자 친구의 친구와 여자 친구의 친구의 새로 만나는 남자를 만났다. 나와 동갑의 그는 상대 여자와 만남 초기이기에 나름 신경쓴 옷차림에 크라이슬러를 끌고 우리를 맞이했다. 음 크라이슬러.. 들어보니 집에 팬션도 있고 사는 집 아이 같다. 아버지가 건축업에서 일한다고 하니.. 음..
오늘 아침부터 일이 손에 잡히지 않는다. 나름 미래 지향적이고 물질적인 것에 많이 연연하지 않는다고 생각했는데 역시나 마찬가지다. 크라이슬러만 생각난다. 내 나름 열심히 살았다고 생각하고 여지껏 걸어온 길의 대부분에 자신감이 있었는데 내 믿음이 흔들린다. 내가 지금 하는 일이 보잘 것 없어보이며 내가 일궈낸(물론 개뿔없지만) 경력들이 의미 없어보인다. 크라이슬러 하나에. ㅋ
몇일전 미국으로 유학은 가는 회사동료가 회식자리에 왔었다. 그때 내 옆에 멍청한 부서장이 나에게 저친구 유학가는거 부럽지 않냐고 물었다. 부럽다라.. 유학을 가는게 부럽냐라니.. 멍청한 질문이다. 예전부터 그 유학가는 회사 동료와 대화를 나눌 때부터 부럽고 부끄러운 점이 있었다. 그는 진정 완고함을 지녔다. 자신의 일에 대해 깊은 믿음을 가지고 있었고 어떠한 망설임이나 선택에 대한 걱정따윈 없는듯 보이는 그 모습. 난 그의 그 모습이 부러웠다. 그 완고함을 가지고 있는것. 그런 정도의 완고함이라면 비록 지금 바닥에 있더라도 결코 그 어느 누구도 부럽지 않고 내 자신의 일 하나하나에 즐거움을 느낄 것이라고 생각된다.
내가 정확하게 유학, 박사를 하겠다는 말을 못하는 이유도 이와 같은 것이 아닐까 한다. 아직 그 완고함이 부족하다. 내겐. 더 깊은 열망과 완고함을 위해 더 진하게 살도록 하겠다.
긍정의 요소가 있어가 어떠한 일에 대해 가능성을 느낄수 있고 열정의 요소가 있어야 그 가능성을 찾으려는 의지가 생기고 완고함이 있어야 발견한 가능성을 실현 할수 있는것 같다.
꼭 필요하다고 느끼는 이 세가지 모두는 내게 그리 많이 주어지지 않았다. 특히나 이번엔 완고함에 대해 이야기 하고자 한다. 머리속에서 지워지지 않은 일이 있으므로..
완고함이라 함은 어찌보면 의지이기도 하고 믿음이기도 하다. 내가 원하는걸 해낼거라는 의지이자 내가 하는 일이 옳은 일이라고 생각하는 믿음.
어제 일이다. 하루종일 까페에 앉아서 내가 원하는 공부를 하고 내가 원하는 논문을 읽고 나름 보람찬 하루를 보낸 후, 늦은 저녁에 여자 친구와 함께 여자 친구의 친구와 여자 친구의 친구의 새로 만나는 남자를 만났다. 나와 동갑의 그는 상대 여자와 만남 초기이기에 나름 신경쓴 옷차림에 크라이슬러를 끌고 우리를 맞이했다. 음 크라이슬러.. 들어보니 집에 팬션도 있고 사는 집 아이 같다. 아버지가 건축업에서 일한다고 하니.. 음..
오늘 아침부터 일이 손에 잡히지 않는다. 나름 미래 지향적이고 물질적인 것에 많이 연연하지 않는다고 생각했는데 역시나 마찬가지다. 크라이슬러만 생각난다. 내 나름 열심히 살았다고 생각하고 여지껏 걸어온 길의 대부분에 자신감이 있었는데 내 믿음이 흔들린다. 내가 지금 하는 일이 보잘 것 없어보이며 내가 일궈낸(물론 개뿔없지만) 경력들이 의미 없어보인다. 크라이슬러 하나에. ㅋ
몇일전 미국으로 유학은 가는 회사동료가 회식자리에 왔었다. 그때 내 옆에 멍청한 부서장이 나에게 저친구 유학가는거 부럽지 않냐고 물었다. 부럽다라.. 유학을 가는게 부럽냐라니.. 멍청한 질문이다. 예전부터 그 유학가는 회사 동료와 대화를 나눌 때부터 부럽고 부끄러운 점이 있었다. 그는 진정 완고함을 지녔다. 자신의 일에 대해 깊은 믿음을 가지고 있었고 어떠한 망설임이나 선택에 대한 걱정따윈 없는듯 보이는 그 모습. 난 그의 그 모습이 부러웠다. 그 완고함을 가지고 있는것. 그런 정도의 완고함이라면 비록 지금 바닥에 있더라도 결코 그 어느 누구도 부럽지 않고 내 자신의 일 하나하나에 즐거움을 느낄 것이라고 생각된다.
내가 정확하게 유학, 박사를 하겠다는 말을 못하는 이유도 이와 같은 것이 아닐까 한다. 아직 그 완고함이 부족하다. 내겐. 더 깊은 열망과 완고함을 위해 더 진하게 살도록 하겠다.
Saturday, July 24, 2010
chapter 5 (TCP 기반 서버 / 클라이언트 2)
앞에서 구현한 TCP 서버/ 클라이언트는 프로그래밍 관점에서만 공부한 것이다. 이는 TCP의 동작 방식을 고려 하지 않았는데 이번장을 통해 TCP의 이론적인 부분을 이해한다.
-에코 클라이언트의 완벽 구현-
 왼쪽 echo_client.c를 보면 45~46의 코드를 보면 "read, write 함수가 호출될 때마다 문자열 단위로 실제 입출력이 이뤄진다"는 잘못된 가정이 있는 것을 알수 있다. 그러나 TCP는 연결 지향형으로 전송 데이터의 경계가 없다. 그렇기 때문에 한 데이터를 서버가 두 패킷에 나눠 보낼수도 있고 여러 문자열이 한 패킷에 올 수도 있다. 이 문제를 아래 echo_client2.c 에서 보듯이 write로 전송한 문자열의 길이만큰 read로 읽을때까지 반복하면 해결된다.
왼쪽 echo_client.c를 보면 45~46의 코드를 보면 "read, write 함수가 호출될 때마다 문자열 단위로 실제 입출력이 이뤄진다"는 잘못된 가정이 있는 것을 알수 있다. 그러나 TCP는 연결 지향형으로 전송 데이터의 경계가 없다. 그렇기 때문에 한 데이터를 서버가 두 패킷에 나눠 보낼수도 있고 여러 문자열이 한 패킷에 올 수도 있다. 이 문제를 아래 echo_client2.c 에서 보듯이 write로 전송한 문자열의 길이만큰 read로 읽을때까지 반복하면 해결된다.

에코 클라이언트 이외의 경우에는? 에플리케이션 프로토콜의 정의 : 위의 경우는 수신할 데이터의 크기를 알 경우지만 일반적으로는 그렇지 않다. 이럴 때 필요한것이 어플리케이션 프로토콜.
어플리케이션 프로토콜이란? 클라이언트의 구현과정에서 만들어지는 약속
-TCP의 이론적인 이야기-
TCP 소켓의 생성에서 소멸과정을 크게 3단계로 나눌수 있다.
1.상대 소켓과의 연결 : Three-way handshaking (3번의 shaking에 걸쳐서 이루어진다. 호스트 A가 접속하고자 하는 호스트 B에 패킷을 보내고 B에서는 받은 패킷을 확인하면서 새로운 패킷을 보내고 다시 A가 B에서 온 패킷을 확인하는 과정)
2.상대 소켓과의 데이터 송수신 :
3.상대 소켓과의 연결 종료 : Four-way handshaking으로 호스트 A가 연결을 끊기 위한 패킷을 보내면 호스트 B에서 응답 패킷을 보내고 B에서 다시 한번 종료 패킷을 보내면 A에서는 그 패킷을 보고 연결을 종료한다.
-에코 클라이언트의 완벽 구현-
 왼쪽 echo_client.c를 보면 45~46의 코드를 보면 "read, write 함수가 호출될 때마다 문자열 단위로 실제 입출력이 이뤄진다"는 잘못된 가정이 있는 것을 알수 있다. 그러나 TCP는 연결 지향형으로 전송 데이터의 경계가 없다. 그렇기 때문에 한 데이터를 서버가 두 패킷에 나눠 보낼수도 있고 여러 문자열이 한 패킷에 올 수도 있다. 이 문제를 아래 echo_client2.c 에서 보듯이 write로 전송한 문자열의 길이만큰 read로 읽을때까지 반복하면 해결된다.
왼쪽 echo_client.c를 보면 45~46의 코드를 보면 "read, write 함수가 호출될 때마다 문자열 단위로 실제 입출력이 이뤄진다"는 잘못된 가정이 있는 것을 알수 있다. 그러나 TCP는 연결 지향형으로 전송 데이터의 경계가 없다. 그렇기 때문에 한 데이터를 서버가 두 패킷에 나눠 보낼수도 있고 여러 문자열이 한 패킷에 올 수도 있다. 이 문제를 아래 echo_client2.c 에서 보듯이 write로 전송한 문자열의 길이만큰 read로 읽을때까지 반복하면 해결된다.
에코 클라이언트 이외의 경우에는? 에플리케이션 프로토콜의 정의 : 위의 경우는 수신할 데이터의 크기를 알 경우지만 일반적으로는 그렇지 않다. 이럴 때 필요한것이 어플리케이션 프로토콜.
어플리케이션 프로토콜이란? 클라이언트의 구현과정에서 만들어지는 약속
-TCP의 이론적인 이야기-
TCP 소켓의 생성에서 소멸과정을 크게 3단계로 나눌수 있다.
1.상대 소켓과의 연결 : Three-way handshaking (3번의 shaking에 걸쳐서 이루어진다. 호스트 A가 접속하고자 하는 호스트 B에 패킷을 보내고 B에서는 받은 패킷을 확인하면서 새로운 패킷을 보내고 다시 A가 B에서 온 패킷을 확인하는 과정)
2.상대 소켓과의 데이터 송수신 :
3.상대 소켓과의 연결 종료 : Four-way handshaking으로 호스트 A가 연결을 끊기 위한 패킷을 보내면 호스트 B에서 응답 패킷을 보내고 B에서 다시 한번 종료 패킷을 보내면 A에서는 그 패킷을 보고 연결을 종료한다.
Thursday, July 22, 2010
6th DNA base, 5-hydroxymethylcytosine
http://www.genomeweb.com/rockefeller-researchers-identify-sixth-nucleotide
Today I found the article above and new epigenetic factor, 5-hydroxymethylcytosine. I am always so late.
Today I found the article above and new epigenetic factor, 5-hydroxymethylcytosine. I am always so late.
Tuesday, July 20, 2010
chapter 4 (TCP 기반 서버/ 클라이언트 1)
앞서 소켓의 생성과 생성된 소켓에 주소 할당을 알아보았다. 이번에는 연결지향형 소켓을 중심으로 데이터 송수신방법에 대해 살펴보도록 한다.
-TCP와 UDP에 대한 이해-
TCP 소켓 : 프로토콜 체계는 IPv4인 PF_INET 이고 데이터 전송 방식은 연결지향형인 SOCK_STREAM인 유일한 소켓.
 TCP/IP 프로토콜 스택 : 오른쪽 그림에서와 같이 4개의 계층으로 나뉘다. 이는 '인터넷 기반의 효율적인 데이터 전송' 이라는 문제를 하나의 큰프로토콜로 해결하려는 것이 아니라 작은 문제로 나눠서 효율적으로 풀기 위한것으로 표준화 작업을 통한 개방형 시스템 설계의 장점을 가진다.
TCP/IP 프로토콜 스택 : 오른쪽 그림에서와 같이 4개의 계층으로 나뉘다. 이는 '인터넷 기반의 효율적인 데이터 전송' 이라는 문제를 하나의 큰프로토콜로 해결하려는 것이 아니라 작은 문제로 나눠서 효율적으로 풀기 위한것으로 표준화 작업을 통한 개방형 시스템 설계의 장점을 가진다.
LINK 계층 : 물리적 영역의 표준화로 LAN과 같은 네트워크 표준과 관련된 프로토콜을 정의하는 영역
IP 계층 : 목적지로 데이터를 전송하기 위해서 어떤 경로를 거쳐갈지를 해결하는 것이 IP 계층이고 이 계층에서 사용하는 프로토콜이 IP(internet protocol) 이다. 비연결지향적이며 데이터 손실이 있을 수 있어 신뢰할만한 프로토콜이 아니다.
TCP/UDP 계층 : IP계층에서 데이터 전송을 위한 경로를 알려주면 이를 바탕으로 실제 데이터 송수신을 담당하는 것이 TCP/UDP 경로이고 TCP가 상대적으로 복잡하다. IP 자체는 신뢰할만하지 않기 때문에 TCP 를 통해서 데이터 전송의 성공 확인을 하면서 안정적인 데이터 전송이 가능해 진다.
APPLICATION 계층 : 위의 과정은 소켓이라는 것 하나에 감춰져 있기에 프로그래머들이 자유로워 지는데 이러한 소켓이라는 도구를 사용하여 프로그램의 성격에 데이터 송수신에 대한 규칙을 APPLICATION 프로토콜이라 한다.
-TCP기반 서버, 클라이언트 구현-
 TCP 서버에서의 기본적인 함수호출 순서 : socket(소켓 생성) -> bind(소켓 주소 할당) -> listen(연결 요청 대기 상태) -> accept(연결허용) -> read/write(데이터 송수신) -> close(연결종료)
TCP 서버에서의 기본적인 함수호출 순서 : socket(소켓 생성) -> bind(소켓 주소 할당) -> listen(연결 요청 대기 상태) -> accept(연결허용) -> read/write(데이터 송수신) -> close(연결종료)
함수의 매개변수와 return 형은 그림 참조
연결요청 대기상태로의 진입 : listen 함수가 호출되어야 클라이언트에 connect 함수가 호출될 수 있다.
매개인자 sock 은 '연결요청 대기상태'(클라이언트가 연결요청을 했을 때 연결 수락까지 요청을 대시시킬수 있는 상태)에 두고자 하는 소켓의 파일 디스크립터, backlog는 '연결 요청 대기 queue'의 크기 정보로 backlog를 5로 하였으면 클라이언트의 연결요청을 5개까지 대기 시킬수 있게 된다.
정리하자면 socket 함수로 생성된 소켓이 listen함수에 의해 문지기 소켓(서버 소켓)이 되어 banklog 만큼의 크기로 대기실을 만들고 이러한 상태가 되면 이를 '연결 요청 대기 상태'라고 한다.
클라이언트의 연결요청 수락 : socket 함수에 의해 만들어진 소켓은 listen함수에 의해 서버소켓이 되고 클라이언트의 연결요청을 대기시킨다. 이러한 대기중인 클라이언트와 데이터를 주고 받을 소켓이 하나 더 필요하다. 이는 accept 함수를 호출함으로서 소켓이 만들어 지고 이 소켓은 대기중인 클라이언트 소켓과 자동으로 연결된다.
accept 함수의 매개변수는 bind 함수와 같으나 주소정보인 addr 은 bind에서 서버 자신의 주소아지만 accept에서는 연결요청 한 클라이언트의 주소정보이다. 또한 마지막 매개변수인 addrlen은 주소의 변수의 크기를 다른 변수에 저장한 뒤 그 변수의 주소 값을 전달해야 한다.
 TCP 클라이언트의 기본적인 함수호출 순서 : 다음은 클라이언트의 구현 순서이다. socket(소켓생성) - > connect(연결요청) -> read/write(데이터 송수신) -> close(연결종료)
TCP 클라이언트의 기본적인 함수호출 순서 : 다음은 클라이언트의 구현 순서이다. socket(소켓생성) - > connect(연결요청) -> read/write(데이터 송수신) -> close(연결종료)
connect의 매개변수 중 sockdf 는 클라이언 소켓의 파일 디스크립터, servaddr은 연결요청을 하고자하는 서버의 주소 정보, addrlen은 주소의 변수 길이로 크기 정보를 변수에 저장한다음 그 변수의 주소를 넘겨야 한다.
connect함수의 return은 서버의 listen함수에 의해 대기 큐에 등록되거나 오류에 의한 연결중단이 되었을때 반환된다.
서버구현시 bind를 통해 소켓에 자기 자신의 IP와 PORT를 할당하였는데 클라이언트 구현시 이점이 생략되어 있다. 이는 connect 함수가 호출될때 운영체제(커널)에 의해 자신에게 할당된 IP와 임의의 PORT를 할당해서 생략되어지는 것이다.
-Iterative 기반의 서버, 클라이언트 구현-
에코서버와 에코 클라이언트를 구현하고자 한다. 에코 서버는 클라이언트가 전송하는 문자열 데이터를 그대로 재전송하는 서버이다.
 Iterative 서버의 구현 : 오른쪽 그림과 같은 서버의 형식이 iterative 서버이며 이는 한번에 하나의 클라이언트에게만 서비스가 가능하다. 동시에 여러 클라이언트를 상대하기 위해서는 프로세스와 쓰레드의 개념을 알아야 한다.
Iterative 서버의 구현 : 오른쪽 그림과 같은 서버의 형식이 iterative 서버이며 이는 한번에 하나의 클라이언트에게만 서비스가 가능하다. 동시에 여러 클라이언트를 상대하기 위해서는 프로세스와 쓰레드의 개념을 알아야 한다.
이부분은 코드 위주이므로 책을 참조한다.
-TCP와 UDP에 대한 이해-
TCP 소켓 : 프로토콜 체계는 IPv4인 PF_INET 이고 데이터 전송 방식은 연결지향형인 SOCK_STREAM인 유일한 소켓.

LINK 계층 : 물리적 영역의 표준화로 LAN과 같은 네트워크 표준과 관련된 프로토콜을 정의하는 영역
IP 계층 : 목적지로 데이터를 전송하기 위해서 어떤 경로를 거쳐갈지를 해결하는 것이 IP 계층이고 이 계층에서 사용하는 프로토콜이 IP(internet protocol) 이다. 비연결지향적이며 데이터 손실이 있을 수 있어 신뢰할만한 프로토콜이 아니다.
TCP/UDP 계층 : IP계층에서 데이터 전송을 위한 경로를 알려주면 이를 바탕으로 실제 데이터 송수신을 담당하는 것이 TCP/UDP 경로이고 TCP가 상대적으로 복잡하다. IP 자체는 신뢰할만하지 않기 때문에 TCP 를 통해서 데이터 전송의 성공 확인을 하면서 안정적인 데이터 전송이 가능해 진다.
APPLICATION 계층 : 위의 과정은 소켓이라는 것 하나에 감춰져 있기에 프로그래머들이 자유로워 지는데 이러한 소켓이라는 도구를 사용하여 프로그램의 성격에 데이터 송수신에 대한 규칙을 APPLICATION 프로토콜이라 한다.
-TCP기반 서버, 클라이언트 구현-

함수의 매개변수와 return 형은 그림 참조
연결요청 대기상태로의 진입 : listen 함수가 호출되어야 클라이언트에 connect 함수가 호출될 수 있다.
매개인자 sock 은 '연결요청 대기상태'(클라이언트가 연결요청을 했을 때 연결 수락까지 요청을 대시시킬수 있는 상태)에 두고자 하는 소켓의 파일 디스크립터, backlog는 '연결 요청 대기 queue'의 크기 정보로 backlog를 5로 하였으면 클라이언트의 연결요청을 5개까지 대기 시킬수 있게 된다.
정리하자면 socket 함수로 생성된 소켓이 listen함수에 의해 문지기 소켓(서버 소켓)이 되어 banklog 만큼의 크기로 대기실을 만들고 이러한 상태가 되면 이를 '연결 요청 대기 상태'라고 한다.
클라이언트의 연결요청 수락 : socket 함수에 의해 만들어진 소켓은 listen함수에 의해 서버소켓이 되고 클라이언트의 연결요청을 대기시킨다. 이러한 대기중인 클라이언트와 데이터를 주고 받을 소켓이 하나 더 필요하다. 이는 accept 함수를 호출함으로서 소켓이 만들어 지고 이 소켓은 대기중인 클라이언트 소켓과 자동으로 연결된다.
accept 함수의 매개변수는 bind 함수와 같으나 주소정보인 addr 은 bind에서 서버 자신의 주소아지만 accept에서는 연결요청 한 클라이언트의 주소정보이다. 또한 마지막 매개변수인 addrlen은 주소의 변수의 크기를 다른 변수에 저장한 뒤 그 변수의 주소 값을 전달해야 한다.

connect의 매개변수 중 sockdf 는 클라이언 소켓의 파일 디스크립터, servaddr은 연결요청을 하고자하는 서버의 주소 정보, addrlen은 주소의 변수 길이로 크기 정보를 변수에 저장한다음 그 변수의 주소를 넘겨야 한다.
connect함수의 return은 서버의 listen함수에 의해 대기 큐에 등록되거나 오류에 의한 연결중단이 되었을때 반환된다.
서버구현시 bind를 통해 소켓에 자기 자신의 IP와 PORT를 할당하였는데 클라이언트 구현시 이점이 생략되어 있다. 이는 connect 함수가 호출될때 운영체제(커널)에 의해 자신에게 할당된 IP와 임의의 PORT를 할당해서 생략되어지는 것이다.
-Iterative 기반의 서버, 클라이언트 구현-
에코서버와 에코 클라이언트를 구현하고자 한다. 에코 서버는 클라이언트가 전송하는 문자열 데이터를 그대로 재전송하는 서버이다.

이부분은 코드 위주이므로 책을 참조한다.
Monday, July 19, 2010
De novo assembly of a 40 Mb Eukaryotic Genome from Short Sequence Reads: Sordaria macrospora, a Model Organism for Fungal Morphogenesis
To see the most recent de novo assembly paper, I found this paper(http://www.plosgenetics.org/article/info:doi/10.1371/journal.pgen.1000891). I will just focus on genomic assembly part in this paper.
They use solexa and FLX data. They produce a single and two paired-end data of different insert size libraries from solexa and single read from FLX. The amount of data showed in figures. A little bit doubtful point is the amount of read is too small compared with my data. This maybe come from the difference of version of solexa.

assembly process
The assembly process is simple. They assembled the total raw data from both system and contig from FLX data with Velvet. They said that FLX data reduced the gaps in contigs and addition of paired-end data improved N50 size.
confirmation of assembly
Checking the existence of synteny block between draft genome and closed species. Predicted protein from draft genome mapped to relatively closed species.
They use solexa and FLX data. They produce a single and two paired-end data of different insert size libraries from solexa and single read from FLX. The amount of data showed in figures. A little bit doubtful point is the amount of read is too small compared with my data. This maybe come from the difference of version of solexa.

assembly process
The assembly process is simple. They assembled the total raw data from both system and contig from FLX data with Velvet. They said that FLX data reduced the gaps in contigs and addition of paired-end data improved N50 size.
confirmation of assembly
Checking the existence of synteny block between draft genome and closed species. Predicted protein from draft genome mapped to relatively closed species.
Genomic Analysis of Organismal Complexity in the Multicellular Green Alga Volvox carteri
I choose this paper for reporting in full, because
1. this is published in science (what I feel in these days is that science is the most hard journal to understand exactly, this means that the paper in this journal compress enormous knowledge, so this make me study a lot),
2. this paper contain de novo assembly, so I want to find how they do that and confirm the assemly,
3. they compare two relative species,
All of these reasons are the factors that make me feel this paper can be my role model paper.
Saturday, July 17, 2010
AHEAD(the Alliance for Human Epigenomics and Disease)
I found from last paper that Epigenome Project is continued by AHEAD. In this post, I will introduce the article about AHEAD which is published in Nature. Ah.. the funny thing is.. when I was googling about AHEAD, I found that Prof, Young-Joon Kim presented his research in AHEAD conference in last year.
The title of article which will be introduced is "Moving AHEAD with an international human epigenome project". This was published on August in 2008.
A plan to 'genomicize' epigenomics research and pave the way for breakthroughs in the prevention, diagnosis and treatment of human disease.
here we discuss the benefits of the AHEAD frame work to coordinate and plan an international Human Epigenome Project.
 Epigenetic mechanisms : histone modification, positioning of histone variants, nucleosome remodelling, DNA methylation, small and non-coding RNAs. These things interact with transcription factor or other protein to regulate target gene expression.
Epigenetic mechanisms : histone modification, positioning of histone variants, nucleosome remodelling, DNA methylation, small and non-coding RNAs. These things interact with transcription factor or other protein to regulate target gene expression.
epigenetic mechanisms are recognized as being involved disease.
although this mechanisms have heritable characteristics, drug can reverse them.
So more comprehensive characterization of them is needed to maximize utility of them in treatment of disease.
the goal of AHEAD project is provide high-resolution reference epigenome maps.
an international project would provide the bioinformatics tool.
-Early steps-
About from 2004 global movements were appeared to organize the international community.
 In Europe, strong tradition for epigenetic study supported by European Union funding programmes or individual national initiatives. more than US $79M was supported to DNA methylation(HEP, Human Epigenome Project), chromatin profiling(HEROIC,High-Throughput Epigenetic Regulatory Organization In Chromatin) and treatment of neoplastic disease(EPITRON, EPIgenetic TReatment Of Neoplastic disease). The special function is provided by the NoE(Epigenome Network of Excellence).
In Europe, strong tradition for epigenetic study supported by European Union funding programmes or individual national initiatives. more than US $79M was supported to DNA methylation(HEP, Human Epigenome Project), chromatin profiling(HEROIC,High-Throughput Epigenetic Regulatory Organization In Chromatin) and treatment of neoplastic disease(EPITRON, EPIgenetic TReatment Of Neoplastic disease). The special function is provided by the NoE(Epigenome Network of Excellence).
In U.S. 2004 NCI(international cancer institute)-sponsored Epigenetic Mechanisms in Cancer Think Tank, 2005 NCI workshop, AACR(The Ametican Association for Cancer Research) organized a Human Epigenome Workshop in 2005, 2006.
On the heels of these workshop AACR Human Epigenome Task Force was formed to design strategy and develop a timetable for the implementation of an international Human Epigenome Project. This task force recommended the formation of AHEAD to coordinate a transdisciplinary, international project.
In summary, each country (EU, US) organize and develop their own community and project. And after Human peigenome workshop from AACR, AACR Human Epigenome Task Force was formed and they made AHEAD, internation project to map a defined subject of robust epigenetic makers and support bioinformatics infrastructure.
-The scope of AHEAD-
1.provide complete epigenome maps at very high resolution for important histone modifications across diverse cellular states in both human and mouse
2.complete and catalogue epigenome maps of model yeasts, plants, and animals
3.deliver a high resolution DNA methylation map of the entire human genome in defined cell types and a landmark map for transcription start sites of all protein coding genes and a representative number of other features throughout the genome
4.define non-coding and small RNAs
5.establish a bioinformatics platform including a relational database, website and suite of analytic tools to organize, intergrate and display whole epigenomic data on model organisms and humans.
AHEAD differ from and complement ENCODE(ENCyclopedia Of DNA Elements).
ENCODE is focused on defining the functional sequences in the genome.
AHEAD define the patterns of epigenetic regulation at that sequences.
-Reference epignomes-
 The criteria for selection of the reference system:
The criteria for selection of the reference system:
1.cells should be easy to sample in a reporoducible fashion.
2.cell numbers should be sufficient for analyses of DNA methylation and chromatin modifications.
3.cell progenitors should be identified that can be suitably manipulated and harvested in a pure state.
4.where possible, cells should be amenable to tissue reconstruction and three-dimensional model systems
5.systems must provide insight into key differentiation and related disease states.

-Advances in technology-
there is nothing new, so I just skip this part
-Model organisms-
-Computational challenges-
A central relational database and a web interface which include analytic and statistical tools to present and visualize data are needed.
The title of article which will be introduced is "Moving AHEAD with an international human epigenome project". This was published on August in 2008.
A plan to 'genomicize' epigenomics research and pave the way for breakthroughs in the prevention, diagnosis and treatment of human disease.
here we discuss the benefits of the AHEAD frame work to coordinate and plan an international Human Epigenome Project.

epigenetic mechanisms are recognized as being involved disease.
although this mechanisms have heritable characteristics, drug can reverse them.
So more comprehensive characterization of them is needed to maximize utility of them in treatment of disease.
the goal of AHEAD project is provide high-resolution reference epigenome maps.
an international project would provide the bioinformatics tool.
-Early steps-
About from 2004 global movements were appeared to organize the international community.

In U.S. 2004 NCI(international cancer institute)-sponsored Epigenetic Mechanisms in Cancer Think Tank, 2005 NCI workshop, AACR(The Ametican Association for Cancer Research) organized a Human Epigenome Workshop in 2005, 2006.
On the heels of these workshop AACR Human Epigenome Task Force was formed to design strategy and develop a timetable for the implementation of an international Human Epigenome Project. This task force recommended the formation of AHEAD to coordinate a transdisciplinary, international project.
In summary, each country (EU, US) organize and develop their own community and project. And after Human peigenome workshop from AACR, AACR Human Epigenome Task Force was formed and they made AHEAD, internation project to map a defined subject of robust epigenetic makers and support bioinformatics infrastructure.
-The scope of AHEAD-
1.provide complete epigenome maps at very high resolution for important histone modifications across diverse cellular states in both human and mouse
2.complete and catalogue epigenome maps of model yeasts, plants, and animals
3.deliver a high resolution DNA methylation map of the entire human genome in defined cell types and a landmark map for transcription start sites of all protein coding genes and a representative number of other features throughout the genome
4.define non-coding and small RNAs
5.establish a bioinformatics platform including a relational database, website and suite of analytic tools to organize, intergrate and display whole epigenomic data on model organisms and humans.
AHEAD differ from and complement ENCODE(ENCyclopedia Of DNA Elements).
ENCODE is focused on defining the functional sequences in the genome.
AHEAD define the patterns of epigenetic regulation at that sequences.
-Reference epignomes-

1.cells should be easy to sample in a reporoducible fashion.
2.cell numbers should be sufficient for analyses of DNA methylation and chromatin modifications.
3.cell progenitors should be identified that can be suitably manipulated and harvested in a pure state.
4.where possible, cells should be amenable to tissue reconstruction and three-dimensional model systems
5.systems must provide insight into key differentiation and related disease states.

-Advances in technology-
there is nothing new, so I just skip this part
-Model organisms-
-Computational challenges-
A central relational database and a web interface which include analytic and statistical tools to present and visualize data are needed.
pseudo gene
I felt the need to clarify about pseudo gene while I organized an annotation of zymo. Therefore in this post I will review about pseudo gene, what it is and how it was happened.
All information comes from http://en.wikipedia.org/wiki/Pseudogene
Pseudogene : non-expressed or defunctional relatives of known gene (homology to a known gene, nonfunctionality).
1.homology to a known gene : usually sequence identity is between 40% and 100%.
2.nonfunctionality : if any one of the common processes, which are needed in making functional product from DNA, such as transcription and pre-mRNA processing fails, this will be nonfunctional. In high-throughput psudogene identification, modification of stop codon and frameshifts are the most common factors.
Types and origin of pseudogenes
1. Processed (retrptransposed) pseudogenes : retrotransposition event is common in mammals and in case of human 30%~44% of genome is composed of this. In retrotransposition, mRNA transcript of a gene is reverse transcribed back into DNA become pseudogene. This kind of pseudogene have features of cDNA like poly-A tail, introns spliced out and lack of promoter.
2. Non-processed pseudogenes : Through gene duplication A copy of gene is made and subsequent mutation make them as pseudogene. This kind of pseudogene have the most feature of genes.
3. Disable genes (unitary pseudogene) : same mechanism (i.e. mutation) which make non-processed pseudogene happened to genes without duplication and became fixed in population.
All information comes from http://en.wikipedia.org/wiki/Pseudogene
Pseudogene : non-expressed or defunctional relatives of known gene (homology to a known gene, nonfunctionality).
1.homology to a known gene : usually sequence identity is between 40% and 100%.
2.nonfunctionality : if any one of the common processes, which are needed in making functional product from DNA, such as transcription and pre-mRNA processing fails, this will be nonfunctional. In high-throughput psudogene identification, modification of stop codon and frameshifts are the most common factors.
Types and origin of pseudogenes
1. Processed (retrptransposed) pseudogenes : retrotransposition event is common in mammals and in case of human 30%~44% of genome is composed of this. In retrotransposition, mRNA transcript of a gene is reverse transcribed back into DNA become pseudogene. This kind of pseudogene have features of cDNA like poly-A tail, introns spliced out and lack of promoter.
2. Non-processed pseudogenes : Through gene duplication A copy of gene is made and subsequent mutation make them as pseudogene. This kind of pseudogene have the most feature of genes.
3. Disable genes (unitary pseudogene) : same mechanism (i.e. mutation) which make non-processed pseudogene happened to genes without duplication and became fixed in population.
DNA methylation profiling of human chromosomes 6, 20 and 22
This paper was found while I have been searching Epignome Project. This is the last paper which was published from Epigenome Project, and I decided to present this on next journal club meeting. This paper have been cited by 317 articles.
The really interesting discovery from introduction of this paper, Epigenome project was dubbed to AHEAD (the Alliance for Human Epigenomics and Disease). And there are some clue (http://www.nature.com/nature/journal/v454/n7205/full/454711a.html, http://www.aacr.org/home/scientists/working-groups--task-forces/task-forces/human-epigenome-task-force.aspx). I will summarize this on next time.
Result

 Distribution of methylation
Distribution of methylation
bimodal distribution of amplicon's methylation state, 27.4% unmethylated(<20% methylation), 42.2 hyper-methylated(>80% methylation) and 30.2 heterogeneously methylated(20%~80%). Only 9.2 of CGIs were hyper-methylated.
 By distinction mosaicism versus imprinting among heterogeneously methylated amplicons, they confirmed the most of them were by mosaicism.
By distinction mosaicism versus imprinting among heterogeneously methylated amplicons, they confirmed the most of them were by mosaicism.
 Comethylation (relationship between the degree of methylation over distance) was quite strong over short distances(<1000 bp). This fact showed that in normal the range of comethylation was shorter than in specific diseases. Absolute difference in methylation betweens tissues were shown in figure. Sperm stood out, and related tissue showed low level of difference.
Comethylation (relationship between the degree of methylation over distance) was quite strong over short distances(<1000 bp). This fact showed that in normal the range of comethylation was shorter than in specific diseases. Absolute difference in methylation betweens tissues were shown in figure. Sperm stood out, and related tissue showed low level of difference.
Promoter methylation
 They divide promoter region in three types. 5' UTR, putative TSSs, Sp1(transcription factor) binding site. And 5' UTR are sub-divided according existence of CGIs, and gene type (novel CDS, novel transcript, pseudo gene).
They divide promoter region in three types. 5' UTR, putative TSSs, Sp1(transcription factor) binding site. And 5' UTR are sub-divided according existence of CGIs, and gene type (novel CDS, novel transcript, pseudo gene).
The most CGIs in 5' UTR are unmethylated, while non CGI 5' UTRs are hyper-methylated. There is no big difference between CDS, transcript, pseudo in exonic region, but in 5' UTR the most amplicon which belong to CDS are unmethylated.
 TSSs showed unmethylated core region of about 1,000 bp.
TSSs showed unmethylated core region of about 1,000 bp.

Age- and sex-dependent DNA methylation
They compared methylation state according to age (26+-4 age, 68+-8 age) and sex, but couldn't find any significant difference.
Differential methylation
T-DMRs (tissue-specific differentially methylated regions) was confirmed by hierarchical clustering which showed biological replicate of each tissue grouped together. 22 % of amplicons were T-DMRs. The frequency of T-DMRs which belong to CGIs was low.
They found that T-DMRs in 5' UTRs which are without CGIs could affect mRNA expression. T-DMRs located in gene have little effect on mRNA expression.
Conservation of DNA methylation
 To check conservation of methylation across species, they compare 59 orthologous amplicons between human and mouse. The majority of profiles were conserved. They extrapolate that 70 % of orthologous loci between human and mouse may have conserved methylation profiles.
To check conservation of methylation across species, they compare 59 orthologous amplicons between human and mouse. The majority of profiles were conserved. They extrapolate that 70 % of orthologous loci between human and mouse may have conserved methylation profiles.
Like most papers in early stage of high-resolution sequencing for methylation state, This paper also just showed landscape of methylation state over large region in chromosomes. There were just arbitrary classification of methylation state and correlation this classification with some specific conditions. There is no absolute rule.
I think that It will be fun that finding orthologous genes which show different methylation state between human and mouse. Are these genes species-specific genes?
The really interesting discovery from introduction of this paper, Epigenome project was dubbed to AHEAD (the Alliance for Human Epigenomics and Disease). And there are some clue (http://www.nature.com/nature/journal/v454/n7205/full/454711a.html, http://www.aacr.org/home/scientists/working-groups--task-forces/task-forces/human-epigenome-task-force.aspx). I will summarize this on next time.
Result

In this paper, they report the methylation profiling of human chromosomes 6, 20 and 22 in 43 samples derived from 12 different normal tissues. They controlled age and sex, which can affect the methylation state, in each samples. 2,524 amplicons with 1.88 M CpG sites on chromosome 6, 20 and 22 that are associated with 873 genes were sequenced by sanger sequencer.

bimodal distribution of amplicon's methylation state, 27.4% unmethylated(<20% methylation), 42.2 hyper-methylated(>80% methylation) and 30.2 heterogeneously methylated(20%~80%). Only 9.2 of CGIs were hyper-methylated.


Promoter methylation

The most CGIs in 5' UTR are unmethylated, while non CGI 5' UTRs are hyper-methylated. There is no big difference between CDS, transcript, pseudo in exonic region, but in 5' UTR the most amplicon which belong to CDS are unmethylated.


Age- and sex-dependent DNA methylation
They compared methylation state according to age (26+-4 age, 68+-8 age) and sex, but couldn't find any significant difference.
Differential methylation
T-DMRs (tissue-specific differentially methylated regions) was confirmed by hierarchical clustering which showed biological replicate of each tissue grouped together. 22 % of amplicons were T-DMRs. The frequency of T-DMRs which belong to CGIs was low.
They found that T-DMRs in 5' UTRs which are without CGIs could affect mRNA expression. T-DMRs located in gene have little effect on mRNA expression.
Conservation of DNA methylation

Like most papers in early stage of high-resolution sequencing for methylation state, This paper also just showed landscape of methylation state over large region in chromosomes. There were just arbitrary classification of methylation state and correlation this classification with some specific conditions. There is no absolute rule.
I think that It will be fun that finding orthologous genes which show different methylation state between human and mouse. Are these genes species-specific genes?
Wednesday, July 14, 2010
biological classification
While I handle NGS data of microbe like Zymomonas and Paenilbacillus, because of name of microbes, I got a thought to study classification of livings. As you know, like our name, they also have last name and middle and first (maybe this is stupid comparison). I thought that if I can know which last name is in long name of microbe and where they belong through that name, that will be very helpful to me to understand more concretely or feel more intimately with them. Being familiar is really important to do something with, right? So start!
- Biological classification -

Biological classification is a form of scientific taxonomy(<->folk taxonomy ; bugs, ducks). 7 ranks are defined by international nomenclature codes.
Carolus Linnaeus's work is a being base of modern classification. What I understand in wikipedia (http://en.wikipedia.org/wiki/Biological_classification) is only that he shorten the name of living.
Since the 1960s, based on Darwinian principle which believe every species have come from common ancestor, cladistic taxonomy has emerged. This arrange taxon based on phylogenetic tree. This will be posted.
So , I found another webpage for bacterial taxonomy and I think this will be more helpful in practical to me (http://www.microbiologybytes.com/iandi/3a.html).
- Bacteria-Basic Facts -
 What are bacteria? lack of organelles, no nucleus, circular DNA chromosome, maybe cell surface is the most complex region.
What are bacteria? lack of organelles, no nucleus, circular DNA chromosome, maybe cell surface is the most complex region.
Gram stain appearances of medically important bacteria
Gram strain (purple) have been used to dye bacteria for identification by microscope. If bacteria only have cell wall made by peptideglycan, it keep the purple color(gram positive). But the bacteria which have extra cell membrane composed of phospholipid lose the purple color(gram negative). In that case, red dye is used for dyeing.
 This site is focused on bacteria of medical importance and the information for naming of bacteria isn't sufficient.
This site is focused on bacteria of medical importance and the information for naming of bacteria isn't sufficient.
Therefore, I found the other web-site (http://www.bacterio.cict.fr/classification.html). I think this will be last destination.
- Classification, taxonomy and systematics of prokaryotes (bacteria) -
- Additional information -
Strain, clone and species : comments on three basic concepts of bacteriology (http://jmm.sgmjournals.org/cgi/reprint/49/5/397.pdf)
- Biological classification -

Biological classification is a form of scientific taxonomy(<->folk taxonomy ; bugs, ducks). 7 ranks are defined by international nomenclature codes.
Carolus Linnaeus's work is a being base of modern classification. What I understand in wikipedia (http://en.wikipedia.org/wiki/Biological_classification) is only that he shorten the name of living.
Since the 1960s, based on Darwinian principle which believe every species have come from common ancestor, cladistic taxonomy has emerged. This arrange taxon based on phylogenetic tree. This will be posted.
So , I found another webpage for bacterial taxonomy and I think this will be more helpful in practical to me (http://www.microbiologybytes.com/iandi/3a.html).
- Bacteria-Basic Facts -
 What are bacteria? lack of organelles, no nucleus, circular DNA chromosome, maybe cell surface is the most complex region.
What are bacteria? lack of organelles, no nucleus, circular DNA chromosome, maybe cell surface is the most complex region.Gram stain appearances of medically important bacteria
Gram strain (purple) have been used to dye bacteria for identification by microscope. If bacteria only have cell wall made by peptideglycan, it keep the purple color(gram positive). But the bacteria which have extra cell membrane composed of phospholipid lose the purple color(gram negative). In that case, red dye is used for dyeing.
 This site is focused on bacteria of medical importance and the information for naming of bacteria isn't sufficient.
This site is focused on bacteria of medical importance and the information for naming of bacteria isn't sufficient.Therefore, I found the other web-site (http://www.bacterio.cict.fr/classification.html). I think this will be last destination.
- Classification, taxonomy and systematics of prokaryotes (bacteria) -
- Additional information -
Strain, clone and species : comments on three basic concepts of bacteriology (http://jmm.sgmjournals.org/cgi/reprint/49/5/397.pdf)
Tuesday, July 13, 2010
chapter 3 (주소체계와 데이터 정렬)
IP : internet protocol, 데이터의 송수신을 위해 컴퓨터에 부여하는 값
PORT 번호 : 프로그램에서 생성되는 소켓을 구분하기 위해 소켓에 부여되는 번호
-소켓에 할당되는 IP주소와 PORT번호-

 인터넷 주소(Internet Address, IP 주소) : IPv4 (4바이트 주소 체계), IPv6 (6 바이트 주소체계), IPv6는 IP가 부족할것을 고려해 만든 체계이나 아직 IPv4가 대세. IPv4는 그림과 같이 A~E 클래스로 분류된다. E 클래스는 일반적이지 않고 예약되어 있다. 그림에서 나오는 네트워크 주소 (network ID) 란 네트워크 구분의 위한 IP주소의 일부. 예를 들어 데이터를 203.211.172.103의 컴퓨터로 전송한다고 할때 우선은 IP주소의 네트워크 ID (여기서는 203.211.172)로 데이터를 보내고 라우터(네트워크 안의 호스트와 외부의 데이터 송수신을 위한 물리적 장치)가 나머지 호스트 ID (여기서는 172)로 데이터를 전송한다.
인터넷 주소(Internet Address, IP 주소) : IPv4 (4바이트 주소 체계), IPv6 (6 바이트 주소체계), IPv6는 IP가 부족할것을 고려해 만든 체계이나 아직 IPv4가 대세. IPv4는 그림과 같이 A~E 클래스로 분류된다. E 클래스는 일반적이지 않고 예약되어 있다. 그림에서 나오는 네트워크 주소 (network ID) 란 네트워크 구분의 위한 IP주소의 일부. 예를 들어 데이터를 203.211.172.103의 컴퓨터로 전송한다고 할때 우선은 IP주소의 네트워크 ID (여기서는 203.211.172)로 데이터를 보내고 라우터(네트워크 안의 호스트와 외부의 데이터 송수신을 위한 물리적 장치)가 나머지 호스트 ID (여기서는 172)로 데이터를 전송한다.
IP 주소의 클래스 구분법 :
클래스 A 의 첫 바이트 범위 0 ~ 127
클래스 B 의 첫 바이트 범위 128 ~ 191
클래스 C 의 첫 바이트 범위 192 ~ 223
소켓의 구분에 활용되는 PORT번호 : IP는 컴퓨터를 구분하기 위한 값이다. 이런 IP를 보고 전달된 데이터는 컴퓨터의 수많은 프로그램 중에 어떤 프로그램에 사용되어야 하는지 구분되어지기 위해 사용되는 것이 PORT번호이다. 이 PORT번호를 통해 운영체제가 해당 소켓에 데이터를 분배한다. 즉 하나의 운영체제 내에서 소켓을 구분하기 위한것이 PORT 번호이다. 16 비트 (0~65535)이다. 중복되어선 안되며 0~1023는 well-known PORT라고 해서 특정 프로그램에 할당되어져 있기에 이를 제외해서 사용한다.
-주소정보의 표현-

IPv4 기반의 주소표현을 위한 구조체 : 주소정보에는 프로토콜 체계 (예: IPv4), IP 주소, PORT 번호가 들어가 있어야 한다.
오른쪽 구조체에 사용된 자료형과 멤버는 책을 참조. 매우 중요
-네트워크 바이트 순서와 인터넷 주소 변환-
이부분의 위의 주소정보표현의 구조체인 sockaddr_in의 멤버 sin_port와 sin_zero를 위한 것이다.
CPU마다 메모리에 데이터를 저장하고 해석하는 방식이 다르다(호스트 바이트 순서).
빅 엔디안 (Big Endian) : 상위 바이트의 값을 작은 번지수에 저장하는 방식
리틀 엔디안 (Little Endian) : 상위 바이트의 값을 큰 번지수에 저장하는 방식, 대표적으로 intel 계열의 CPU
그래서 CPU마다 다른 방식으로 데이터 저장 방식(호트스 바이트 순서)이 다르므로 이를 통일하고 한것이 "네트워크 바이트 순서"로 빅 엔디안을 뜻한다.
바이트 순서를 변환해주는 함수:
unsigned short htons(unsigned short);
unsigned short ntons(unsigned short);
unsigned long htonl(unsigned long);
unsigned long ntonl(unsigned long);
htons 에서 h는 host를 s는 short를 의미, 즉 htons(h to n s)는 PORT번호(short형 데이터) 를 호스트 바이트 순서에서 네트워크 바이트 순서로 변환하라는 의미
-인터넷 주소의 초기화와 할당-
다음은 코드 설명이 많으므로 책 참조
문자열 정보를 네트워크 바이트 순서의 정수로 변환하기 : 우리가 익숙한 IP 주소는 점이 찍힌 십진수 표현 방식 (Dotted-Decimal Notation)으로 이를 32비트 정수형으로변환해주는 함수가 필요한데 이것이 inet_addr와 inet_aton이다. 차이점은 inet_addr는 성공시 in_addr_t (unsigned 32 bit int)를 실패시 INADDR_NONE을 반환하는 반면, inet_aton은 성공시 1을 실패시 0을 반환하고 매개변수로 구조체인 in_addr의 주소를 받아서 성공시 그곳에 변환된 값을 넣는다.
inet_aton과 반대 역활, 그러니까 32비트 정수형으로 되어 있는 IP를 Dotted-Decimal Notation으로 변환해 주는 함수가 inet_ntoa이다.
인터넷 주소의 초기화 : 아래 그림 참조

클라이언트의 주소정보 초기화 : 클라이언트의 connet 함수 역시 구조체 sockaddr_in를 사용하나 IP와 PORT의 대상이 다르다. 서버 bind 함수의 경우 서버의 IP와 PORT를, 클라이언트의 connect 함수의 경우 서버의 IP와 PORT를 넣어야 한다.
서버의 경우 "IP 211.217.168.13 PORT 9190으로 들어오는 데이터는 내게 보내라"
클라이언트의 경우 "IP 211.217.168.13 PORT 9190으로 연결해라" 라는 의미
INADDR_ANY : 위에서 처럼 매번 bind 함수에 IP를 넣는 것이 귀찮기 때문에 자동화로 IP를 받을 수 있다. 위에 그림에서 serv_ip를 선언하지 말고 addr.sin_addr.s_addr=inet_addr(serv_ip); 대신
addr.sin_addr.s_addr=htonl(INADDR_ANY); 라고 하면 자동으로 IP가 할당된다.
서버소켓 초기화 과정 : 소켓선언과 함께 위 내용을 최종 정리하면 다음 그림과 같다(1장에서 3장까지의 내용 총괄).

PORT 번호 : 프로그램에서 생성되는 소켓을 구분하기 위해 소켓에 부여되는 번호
-소켓에 할당되는 IP주소와 PORT번호-

 인터넷 주소(Internet Address, IP 주소) : IPv4 (4바이트 주소 체계), IPv6 (6 바이트 주소체계), IPv6는 IP가 부족할것을 고려해 만든 체계이나 아직 IPv4가 대세. IPv4는 그림과 같이 A~E 클래스로 분류된다. E 클래스는 일반적이지 않고 예약되어 있다. 그림에서 나오는 네트워크 주소 (network ID) 란 네트워크 구분의 위한 IP주소의 일부. 예를 들어 데이터를 203.211.172.103의 컴퓨터로 전송한다고 할때 우선은 IP주소의 네트워크 ID (여기서는 203.211.172)로 데이터를 보내고 라우터(네트워크 안의 호스트와 외부의 데이터 송수신을 위한 물리적 장치)가 나머지 호스트 ID (여기서는 172)로 데이터를 전송한다.
인터넷 주소(Internet Address, IP 주소) : IPv4 (4바이트 주소 체계), IPv6 (6 바이트 주소체계), IPv6는 IP가 부족할것을 고려해 만든 체계이나 아직 IPv4가 대세. IPv4는 그림과 같이 A~E 클래스로 분류된다. E 클래스는 일반적이지 않고 예약되어 있다. 그림에서 나오는 네트워크 주소 (network ID) 란 네트워크 구분의 위한 IP주소의 일부. 예를 들어 데이터를 203.211.172.103의 컴퓨터로 전송한다고 할때 우선은 IP주소의 네트워크 ID (여기서는 203.211.172)로 데이터를 보내고 라우터(네트워크 안의 호스트와 외부의 데이터 송수신을 위한 물리적 장치)가 나머지 호스트 ID (여기서는 172)로 데이터를 전송한다.IP 주소의 클래스 구분법 :
클래스 A 의 첫 바이트 범위 0 ~ 127
클래스 B 의 첫 바이트 범위 128 ~ 191
클래스 C 의 첫 바이트 범위 192 ~ 223
소켓의 구분에 활용되는 PORT번호 : IP는 컴퓨터를 구분하기 위한 값이다. 이런 IP를 보고 전달된 데이터는 컴퓨터의 수많은 프로그램 중에 어떤 프로그램에 사용되어야 하는지 구분되어지기 위해 사용되는 것이 PORT번호이다. 이 PORT번호를 통해 운영체제가 해당 소켓에 데이터를 분배한다. 즉 하나의 운영체제 내에서 소켓을 구분하기 위한것이 PORT 번호이다. 16 비트 (0~65535)이다. 중복되어선 안되며 0~1023는 well-known PORT라고 해서 특정 프로그램에 할당되어져 있기에 이를 제외해서 사용한다.
-주소정보의 표현-

IPv4 기반의 주소표현을 위한 구조체 : 주소정보에는 프로토콜 체계 (예: IPv4), IP 주소, PORT 번호가 들어가 있어야 한다.
오른쪽 구조체에 사용된 자료형과 멤버는 책을 참조. 매우 중요
-네트워크 바이트 순서와 인터넷 주소 변환-
이부분의 위의 주소정보표현의 구조체인 sockaddr_in의 멤버 sin_port와 sin_zero를 위한 것이다.
CPU마다 메모리에 데이터를 저장하고 해석하는 방식이 다르다(호스트 바이트 순서).
빅 엔디안 (Big Endian) : 상위 바이트의 값을 작은 번지수에 저장하는 방식
리틀 엔디안 (Little Endian) : 상위 바이트의 값을 큰 번지수에 저장하는 방식, 대표적으로 intel 계열의 CPU
그래서 CPU마다 다른 방식으로 데이터 저장 방식(호트스 바이트 순서)이 다르므로 이를 통일하고 한것이 "네트워크 바이트 순서"로 빅 엔디안을 뜻한다.
바이트 순서를 변환해주는 함수:
unsigned short htons(unsigned short);
unsigned short ntons(unsigned short);
unsigned long htonl(unsigned long);
unsigned long ntonl(unsigned long);
htons 에서 h는 host를 s는 short를 의미, 즉 htons(h to n s)는 PORT번호(short형 데이터) 를 호스트 바이트 순서에서 네트워크 바이트 순서로 변환하라는 의미
-인터넷 주소의 초기화와 할당-
다음은 코드 설명이 많으므로 책 참조
문자열 정보를 네트워크 바이트 순서의 정수로 변환하기 : 우리가 익숙한 IP 주소는 점이 찍힌 십진수 표현 방식 (Dotted-Decimal Notation)으로 이를 32비트 정수형으로변환해주는 함수가 필요한데 이것이 inet_addr와 inet_aton이다. 차이점은 inet_addr는 성공시 in_addr_t (unsigned 32 bit int)를 실패시 INADDR_NONE을 반환하는 반면, inet_aton은 성공시 1을 실패시 0을 반환하고 매개변수로 구조체인 in_addr의 주소를 받아서 성공시 그곳에 변환된 값을 넣는다.
inet_aton과 반대 역활, 그러니까 32비트 정수형으로 되어 있는 IP를 Dotted-Decimal Notation으로 변환해 주는 함수가 inet_ntoa이다.
인터넷 주소의 초기화 : 아래 그림 참조

클라이언트의 주소정보 초기화 : 클라이언트의 connet 함수 역시 구조체 sockaddr_in를 사용하나 IP와 PORT의 대상이 다르다. 서버 bind 함수의 경우 서버의 IP와 PORT를, 클라이언트의 connect 함수의 경우 서버의 IP와 PORT를 넣어야 한다.
서버의 경우 "IP 211.217.168.13 PORT 9190으로 들어오는 데이터는 내게 보내라"
클라이언트의 경우 "IP 211.217.168.13 PORT 9190으로 연결해라" 라는 의미
INADDR_ANY : 위에서 처럼 매번 bind 함수에 IP를 넣는 것이 귀찮기 때문에 자동화로 IP를 받을 수 있다. 위에 그림에서 serv_ip를 선언하지 말고 addr.sin_addr.s_addr=inet_addr(serv_ip); 대신
addr.sin_addr.s_addr=htonl(INADDR_ANY); 라고 하면 자동으로 IP가 할당된다.
서버소켓 초기화 과정 : 소켓선언과 함께 위 내용을 최종 정리하면 다음 그림과 같다(1장에서 3장까지의 내용 총괄).

chapter 2 (소켓의 타입과 프로토콜의 설정)
-소켓의 프로토콜과 그에 따른 데이터 전송 특성-
프로토콜(protocol)이란?
컴퓨터 상호간의 대화에 필요한 통신 규약... 너무 식상한 답변이다. 와닿지 않는다.
 소켓(socket) 함수?
소켓(socket) 함수?
인자로 domain (소켓이 사용할 프로토콜 체계 정보 전달), type (소켓의 데이터 전송 방식에 대한 정보 전달), protocol (두 컴퓨터 간 통신에 사용되는 프로토콜 정보 전달) 을 받고 호출 성공시 파일 디스크립터, 실패시 -1 return.
프로토콜 체계(protocol family)? PF_INET, PF_INET6, PF_LOCAL, PF_PACKET, PF_IPX (자세한건 책 참조)
소켓의 타입(type)? 소켓의 데이터 전달 방식. 프로토콜 체계가 정해졌다고 해도 그 안에서 둘 이상의 전송 방식이 있으므로 선택해야 한다. 대표적으로 연결지향형 소켓과 비 연결지향형 소켓.
1. 연결지향형 소켓(SOCK_STREAM) : 독립된 별도의 라인을 통해서 데이터를 전달하는 방식으로 라인상 문제가 없다면; 중간에 데이터 소멸이 없다. 전송 순서대로 데이터가 수신된다. 전송 데이터의 경계가 없다. 소켓 대 소켓의 연결은 반드시 1 대 1이어야 한다.
2. 비 연결지향형 소켓(SOCK_DGRAM) : 전송 순서에 상관없이 가장 빠른 전송을 지향한다. 전송되는 데이터의 손실 및 파손의 우려가 있다. 데이터의 경계가 존재한다. 한번에 전송하는 데이터의 크기가 제한된다.
*데이터 경계가 있다 없다는 데이터 전송을 두번했으면 수신도 두번해야 할경우 경계가 있는것이고 전송 횟수에 상관없이 수신횟수가 있는것을 경계가 없다고 한다.
프로토콜의 최종 선택? 소켓 함수의 세번째 인자.이미 첫번째와 두번째 인자로 충분하기에 세번째 인자가 0이어도 상관이 없으니 특별한 경우 세번째 인자가 필요하다.
"하나의 프로토콜 체계 안에 데이터의 전송 방식이 동일한 프로토콜이 둘 이상 존재할 경우" 전송 방식이 동일한 프로토콜일 경우 이를 좀 더 구체화 해야 한다.
TCP 소켓 : 프로토콜 체계는 IPv4인 PF_INET 이고 데이터 전송 방식은 연결지향형인 SOCK_STREAM 인 유일한 소켓.
UDP 소켓 : PF_INET 이고 SOCK_DGRAM 인 유일한 소켓.
프로토콜(protocol)이란?
컴퓨터 상호간의 대화에 필요한 통신 규약... 너무 식상한 답변이다. 와닿지 않는다.

인자로 domain (소켓이 사용할 프로토콜 체계 정보 전달), type (소켓의 데이터 전송 방식에 대한 정보 전달), protocol (두 컴퓨터 간 통신에 사용되는 프로토콜 정보 전달) 을 받고 호출 성공시 파일 디스크립터, 실패시 -1 return.
프로토콜 체계(protocol family)? PF_INET, PF_INET6, PF_LOCAL, PF_PACKET, PF_IPX (자세한건 책 참조)
소켓의 타입(type)? 소켓의 데이터 전달 방식. 프로토콜 체계가 정해졌다고 해도 그 안에서 둘 이상의 전송 방식이 있으므로 선택해야 한다. 대표적으로 연결지향형 소켓과 비 연결지향형 소켓.
1. 연결지향형 소켓(SOCK_STREAM) : 독립된 별도의 라인을 통해서 데이터를 전달하는 방식으로 라인상 문제가 없다면; 중간에 데이터 소멸이 없다. 전송 순서대로 데이터가 수신된다. 전송 데이터의 경계가 없다. 소켓 대 소켓의 연결은 반드시 1 대 1이어야 한다.
2. 비 연결지향형 소켓(SOCK_DGRAM) : 전송 순서에 상관없이 가장 빠른 전송을 지향한다. 전송되는 데이터의 손실 및 파손의 우려가 있다. 데이터의 경계가 존재한다. 한번에 전송하는 데이터의 크기가 제한된다.
*데이터 경계가 있다 없다는 데이터 전송을 두번했으면 수신도 두번해야 할경우 경계가 있는것이고 전송 횟수에 상관없이 수신횟수가 있는것을 경계가 없다고 한다.
프로토콜의 최종 선택? 소켓 함수의 세번째 인자.이미 첫번째와 두번째 인자로 충분하기에 세번째 인자가 0이어도 상관이 없으니 특별한 경우 세번째 인자가 필요하다.
"하나의 프로토콜 체계 안에 데이터의 전송 방식이 동일한 프로토콜이 둘 이상 존재할 경우" 전송 방식이 동일한 프로토콜일 경우 이를 좀 더 구체화 해야 한다.
TCP 소켓 : 프로토콜 체계는 IPv4인 PF_INET 이고 데이터 전송 방식은 연결지향형인 SOCK_STREAM 인 유일한 소켓.
UDP 소켓 : PF_INET 이고 SOCK_DGRAM 인 유일한 소켓.
chapter 1 (네트워크 프로그래밍과 소켓의 이해)

연결요청을 수락하는 기능의 프로그램을 가르켜 '서버' 라고 하며 기본적으로 다음과 같은 소켓의 생성 과정을 갖는다.
1.소켓 생성 (socket)
2.IP 주소와 PORT번호 할당 (bind)
3.연결요청 가능상태로 변경(listen)
4.연결요청에 대한 수락(accept)
연결요청을 진행하는 프로그램을 가르켜 '클라이언트'라고 이 클라이언트 소켓은 connect 함수로 생성한다.
리눅스는 소켓을 파일의 일종으로 구분한다. 반면 윈도우는 파일과 소켓을 구분하기에 별도의 데이터 송수신 함수를 참조해야 한다 (윈도우 내용은 생략한다).
리눅스에서 소켓을 파일의 일종으로 취급하기에 저수준 파일 입출력 함수(저수준이란 ANSI 표준에서 정의한 함수가 아닌 운영체제가 독립적으로 제공하는 함수)인 open, close, write, read 함수로 처리가 된다.
-파일 디스크립터 : 스트림에 할당한 번호로 운영체제에서 스트림을 편하게 구분하기 위해 붙인 숫자. 윈도우에서는 '파일 핸들'이라고도 한다.
Monday, July 12, 2010
synthetic biology
Synthetic biology is a big issue in these days. Craig Venter, George Church, and Kristala Prather who are the most famous in biology field spoke about synthetic biology to a presidential commission(http://www.genomeweb.com//node/944858?hq_e=el&hq_m=762062&hq_l=4&hq_v=37b0a0cbd3). Then what is synthetic biology? Actually what I know is very little thing. So I make opportunity to know this concept.
Most of reference come from http://2009.igem.org/wiki/images/0/0d/Sins,_Ethics_and_Biology.pdf.
In 1912, the French chemist Stephane Leduc was first to use the term 'synthetic biology'.
In 1978, Szybalski and Skalka quote the term as closer meaning in today.

top-down approach
bottom-up approach
engineering principles considered in synthetic biology, In 2005 Drew Endy's paper are
standardization, decoupling, abstraction.
application of synthetic biology, example is artemisinin, a drug against malaria produced from plant, can be produced in genetically engineer yeasts.

Although there are big differences in synthetic biology(more normalized, standardized and concrete), I accept the concept of synthetic biology as one of the way to comprehend the biology. From the past scientist have tried to modify organism for interrogating biological knowledge and producing what they want more efficiently. Synthetic biology is a part of this movement.
Here are articles published from JCVI(J. Craig Venter Institute).
http://www.sciencemag.org/cgi/content/abstract/1173759
http://www.sciencemag.org/cgi/content/full/329/5987/52
Sunday, July 11, 2010
human epigenome project
As I try to study epigenomics, investigating global movement such as epigenome project to check available data is also important as much as studying biological investigation. And in that sense, I will introduce human epigenome project.
In 2003, scientists from UK's Sanger Institute and Epigenomics, Inc formed the Human Epigenome Project (http://science.howstuffworks.com/genetic-science/human-epigenome-project1.htm). According to website ( http://www.epigenome.org/index.php), The human epigenome project (HEP) is a collaboration run by the members in the Human Epigenome Consortium. One of the aims of the HEP is to generate tissue-specific DNA methylation reference profiles of the human genome. They use sodium bisulfite treatment, find MVPs (methylation variable positions) and release these data.
So far, two papers were published from here.
The first paper is "DNA methylation profiling of the human major histocompatibility complex: a pilot study for the human epigenome project" (http://www.ncbi.nlm.nih.gov/pubmed/15550986?dopt=Abstract)
and the second one is "DNA methylation profiling of human chromosomes 6, 20 and 22"
releasing date from this is stopped in 2006. I am not sure whether their activities stopped.
Monday, July 5, 2010
essential utility (make)
make는 파일 관리 유틸리티이다. 파일 간 종속 관계를 파악하여 기술 파일(Makefile)에 기술된 대로 컴파일 명령이라던지 쉘 명령을 내린다.
make
make는 기술 파일(makefile)에 내용대로 명령을 수행하므로 makefile 작성법과 make 특징만 알면 된다.
-make의 기본 동작-
Sunday, July 4, 2010
essential utility (gcc)
컴파일의 의미와 과정, 그리고 binutils 패키지 활용 방법에 대해 알아본다.
gcc
-컴파일의 의미-
컴파일이란? 형식언어(C, C++) 를 기계어로 번역하는 것.
그럼 기계어(machine instruction)란? 이진수로 되어 있는 숫자로써 CPU의 종류마다 고유하며, 특정 행동을 취하게 하기 위한 코드 (예로 01010101은 i386 CPU에서 'ebp레지스트를 현재의 스택에 push 해라'). 어셈블리 언어와 1대 1 매치.
그럼 어셈블리 언어란? 기계어를 사람이 일상 생활에서 사용하는 자연어에 가깝게 1~6개의 문자로 기호화해서 나타낸 것.
C 소스 파일 컴파일시 -g 옵션을 넣어주고 컴파일 한뒤에 실행 파일을 objdump 명령어로 보면 디버깅 심벌과 기계어, 그리고 어셈블리 코드를 볼수 있다. 기계어는 사람이 보기 좋게 16진수로 되어 있다 (gcc -g -o like like.c --> objdump -S like).
책에 CPU의 0과 1값을 받는 것에 대한 설명이 그림과 함께 잘 나타나 있으나 생략한다.
-C 소스 컴파일 과정-

gcc 는 GNU에서 만든 C 컴파일러이다 (사실 C, Fortran, ada 등도 컴파일 가능하다). 실제로 gcc는 실제 컴파일을 하는 것이 아니라 전처리기(cpp0), C 컴파일러(cc1), 어셈블러(as), 링커(ld 혹은 collect2)를 호출하는 역할을 한다 (*오른쪽 그림 참조*).
컴파일 과정을 gcc -v --save-temps 으로 확인할 수 있다 (-v 는 컴파일 과정을 화면에 출력하라, --save-temps는 컴파일 과정의 파일을 지우지 마라 라는 의미). 위 결과 전처리 후 생기는 *.i 파일 등을 이용하여 에러 수정에 도움이 된다.
1.cpp0에 의한 전처리 과정: /usr/lib/gcc/x86_64-redhat-linux/3.4.6/specs에는 gcc가 호출하는 프로그램의 기본적인 옵션들이 들어 있다. 전처리 과정은 크게 헤더 파일 삽입과 매트로의 치환및 적용으로 이루어진다. 헤더파일 삽입은 include 한 자리에 파일의 내용이 붙게 된다. 매크로 치환은 define으로 된 내용을 찾아 치환이 일어난다.

2.cc1에 의한 어셈블리 소스 파일로 컴파일 :
1.어휘분석 : 스캐너가 소스를 토큰(문법적으로 의미있는 최소 단위)으로 나눔
2.구문분석 : 파서가 토큰을 받아서 문법적 오류가 있는지 확인
3.의미분석 : 의미상 오류 검사(변수 선언검사, 자료형 불일치 검사등등).
4.중간언어생성 : 이해 안됨
5.최적화 : 중간 코드 최적화(공통부분식 제거, 연산 강도 경감, 상수 계산. 예, var=5+3을 var=8로바꿈)와 목적 코드 최적화(최대한 레지스터를 사용)로 나뉨.
6.목적코드생성 : 목적코드란 어셈블리 언어를 뜻한다.
3.as에 의한 기계어 코드 생성 : as에 의해서, cc1에 의해 생긴 *.s 파일의 기계어와 데이터가 들어있는*.o 파일을 생성한다. *.o 파일은 ELF 바이너리 포멧(바이너리 파일의 구조를 결정짓는 규약)의 바이너리 파일이다. readelf -a *.o 를 통해 ELF 포멧 바이너리의 정보를 알 수 있다. xxd like.o 명령으로도 전체를 볼 수 있다.
4.collect2에 의한 링킹 과정 : printf와 같은 함수를 사용하기 위해 정적 라이브러리 내지는 공유 라이브러리를 ELF 포멧의 바이너리 파일과 링크 시키다.
정적라이브러리란? 미리 만들어진 함수나 변수들의 묶음으로 미리 컴파일 된 *.o 파일이 하나의 파일 lib[라이브러리 이름].a 형식으로 묶여있는 형태를 뜻한다. 이렇게 묶인 오브젝트들은 컴파일 과정에서 각 바이너리와 링크되어 삽입된다. 그러나 이렇게 되면 링크가 중복되고 파일크기도 커져 공유라이브러리를 사용한다.
공유라이브러리(동적라이브러리)란? 설명이 굉장히 복잡하기에 대략적으로 설명하기로 한다. 공유 라이브러리가 존재하면 링크 과정에서 정적라이브러리 링크 하듯이 하지 않고 단순히 오브젝트 파일에 공유라이브러리를 사용하겠다고 표시만 해놓게 된다. 컴파일이 마쳐져서 프로그램이 실행되어지면 공유라이브러리를 사용한다는 표시를 보고 프로그램과 함께 동적 링크도 같이 로드하고 동적 링크는 로드된 동적 라이브러라와 프로그램 상의 라이브러리를 매핑한다.
책에는 gcc에서 호출하는 프로그램들(cpp0, cc1, as, collect2)의 옵션설명이 자세히 나와있으며 바이너리를 조작하거나 정보를 보기 위한 바이너리 유틸리티들에 대한 설명이 있으나 이는 생략한다.
Finding the fifth base : Genome-wide sequencing of cytosine methylation
This article introduced technical methodology, using next-generation sequencing, for a study on cytosine methylation (http://signal.salk.edu/publications/Lister_Ecker.pdf).
Ryan Lister and Joseph R. Ecker*
*SALK INSTITUTE (http://www.salk.edu/)
The Mammalian Epigenome
As according to UCSC's web site for bioinformatics course, they discriminate bioinformatics and computational biology. Of course, discrimination of these things would be meaningless because boundary between them is vague and we cannot weigh up the computational skill and the biological knowledge. Maybe this is only my own problem, my effort as a bioinformatician is leaning toward to raise computational skill. Even in the recent situation where contents is emphasizing, lack of biological insight can be serious problem for being leader of the field which I belong. Thus I planned to make group with lab members, who are related with my ex-firm, for studying epigenomics. I ask them to be active on this meeting, and I hope that this meeting will be more than studying groups. This is the first posting for epigenomics.





Bradley E. Bernstein, Alexander Meissner, and Eric S.Lander
-Epigenetic modifications in mammalian genomes
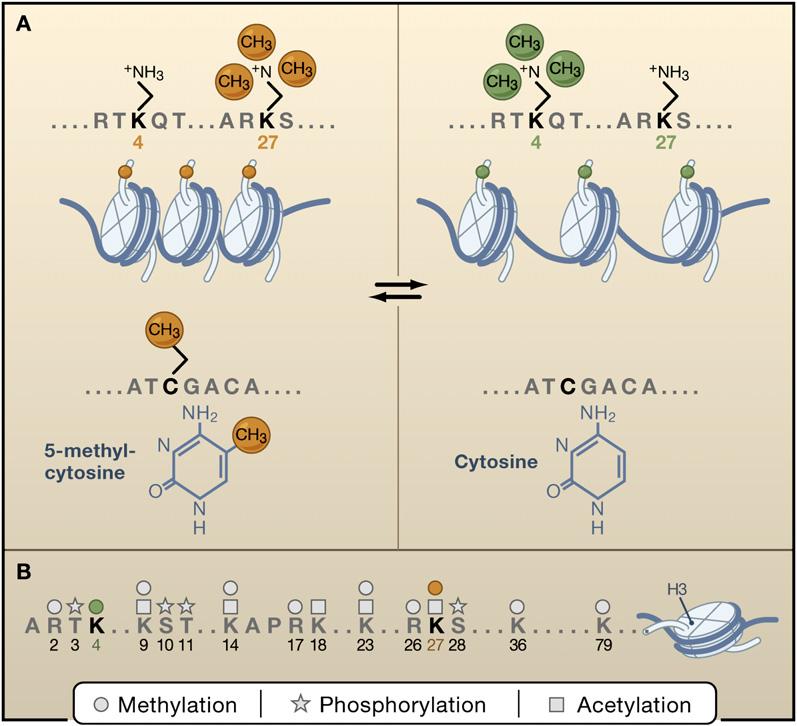
Epigenetic modifications fall into two categories
DNA methylation : In vertebrate, DNA methylation occurs almost exclusively in CpG and most CpG are methylated.
Histone modification : The core histones are subject to more than 100 posttranslational modification (acetylation, methylation, phosphorylation and ubiquitination). This occurs primarily in N-terminal. Lysine acetylation almost correlate to chromatin accessiblity and transcriptional activity, but lysine methylation have various effects (H3K4, H3K36 -> transcribed chromatin, H3K9, H3K27, H4K20 -> repression).
--Epigenetic Inheritance
Epigenetic modifications are heritable. By methyl-transferase Dnmt1 which can methylate hemi-methylated CpG, newly synthesized DNA strand are methylated and the pattern are preserved.
Histone modifications are a little bit hard to explain and need more reference. so I will defer this to next posting.
-A Dynamic Landscape of Cytosine Methylation DNA Methylation
--DNA Methylation in Development and Disease
DNA methylation work as
1.accelerator of recruitment of regulatory protein : methyl-CpG binding proteins recognize methylated cytosine and mediate transcriptional repression through interactions with histone deacetylases.
2.repressor for protein binding to DNA : CTCF binding at the H19 locus.
In development
In cancer, genome-wide loss or aberrant local gain of methylation. Tumor suppressor gene promoter is the target of the latter cases.
--CpG Islands

CpG island cover 0.7 % of genome and contain 7 % of total CpG. In the germ line, CpG in island are unmethylated, so can be recognized and corrected by repair system(deamination of cytosine(uracil), deamination of methylated cytosine(thymine), uracil can be corrected). 60% of promoter are related to CpG islands. The subset of these are tissue specific methylated during development. Some CpG reside within repetitive elements, and these are heavily methylated in somatic tissues. Oct-4 and Nanog promoter's methylation are associated with transcriptional activation while IL-2 and Sry's demethylation are associated.
--Studying the DNA Methylome
bisulfite sequencing : treat DNA with NaHSO3 (this convert unmethylated cytosine to uracil) and then sequence the DNA(uracil read as thymine, methylated cytosine read as cytosine).With this method survey on 6,20,22 chromosome found that small fraction(9.2%) of 511 CpG island are methylated while almost 50 % of non-CpG islands containing 5' UTRs were hypermethylated, and about 70 % conserved methylation profile between mouse and human.
Using fractionation of methylated and unmethylated portions by restriction or antibody : some found that the average methylation level CpG(selected from 5' regions of 371 genes) wa about 35 %. some group detect novel methyaltion at 135 promoters(of which 127 contain CpG islands) when comparing colon cell line with normal colon.
-Global Insights into Histone Biology
--Methods for Large-Scale Analysis of Histone Modifications
--Chromatin Domains and Cellular Memory
--Epigenetic Mechanisms of Genome Plasticity
-The Relationship between Genome and Epigenome

--The Newly Reprogrammed Epigenetic State
ES cells from inner cell mass where remethylation begins in early development, are representative of the reprogrammed state. The role of DNA in defining ES cell epigenome is significant.
H3K4 methylation coincide with CpG islands : trxG complexes that catalyze H3K4 methylation contain domains that bind unmethylated CpG dinucleotide.
H3K27 methylation correlates with transposon-free regions : 1.transposition is incompatible with the chromatin structure or negative slection. OR 2. transposons could interfere with the spreading of H3K27 methylation.
--Repetitive Elements
Transposon-derived DNA sequence is typically highly methylated in somatic tissues. Within given repeat class, modification status appeared to depend on the cellular differentiation state.
--Predicting Cytosine Methylation from DNA Sequences
-Emerging Technologies in Epigenome Research
--Cytosine Methylation
1.MSREs (methylation-sensitive restriction enzymes) , but has limitation on outside the restriction site.
2.Immunoprecipitation with a methylcytosine antibody, also has limitation on resolution
3.bisulfite-treat and sequence analysis, has little bias
--Histone Modifications

Chromatin IP (XChIP) : cross-link DNA and nearby protein by formaldehyde, sonicating the chromatin and then immunoprecipitating with antibody against a particular histone modification. but this has limitations, which are efficacy of antibody, fragmentation bias, and "masked" by other proteins specific to histone.
Native ChIP (N-ChIP) : shearing chromatin by micrococcal nuclease digestion which cut DNA at the length of the linker instead of sonication and no cross-link.
Biotin-tag affinity purification,

DamID (DNA adenine methltransferase identification) : using fusion protein consisting of E.coli Dam(Dam methylates adenines in the sequence GATC) and a certain chromatin protein or TF.
-Future Perspectives and Challenges
There are important issues
1.reduction of the numer of cells required for histone modification studies.
2.better antibody reagents for increasing sensitivity.
3.development of highly parallel DNA analyses.
4.computational tools in need.
I just want to summarize and rearrange the article in my way, but the most of sentences are compressed because of characteristic of review paper, so I couldn't do that.
further study
epigenome project, genomic imprinting & DNA methylation, inheritance of epigenome.
Saturday, July 3, 2010
essential utility (vi)
c 언어를 공부하다가 컴파일을 하게 되고 컴파일을 하다보니 자연스레 make 파일을 알게 되고.. 이와 같은 이유로 좀 더 깊게 알고 싶은 생각에 "유닉스, 리눅스 프로그래밍 필수 유틸리티 (백창우 저)" 를 보기 시작했다. 책은 내용은 프로그래밍을 시작하기 위한 편집기에서부터 컴파일의 자동화를 위한 유틸리티, 디버깅, 그룹 프로그래밍시 프로그램 에디팅 할 때의 버젼 업관리 까지 실직적으로 프로그래밍 시작부터 마무리까지에 필요한 유틸리티를 자세하게 설명하고 있다. 책의 내용이 상당히 많은 관계로 여기서는 가장 기본적인 컨셉과 나에게 도움이 될만한 예를 기록하는것으로 하겠다.
vim 은 3가지 모드로 구성된다.

 붙여넣기를 이해하기 위해서는 레지스트를 알아야 한다. vi 는 총 17개의 레지스트를 갖고 있으며 이를 보기 위해서는 ex모드에서 :reg 명령을 넣으면 된다. 명령모드에서 "1p를 누르게 되면 1번 레지스트에 있는 내용을 붙이게 된다.
붙여넣기를 이해하기 위해서는 레지스트를 알아야 한다. vi 는 총 17개의 레지스트를 갖고 있으며 이를 보기 위해서는 ex모드에서 :reg 명령을 넣으면 된다. 명령모드에서 "1p를 누르게 되면 1번 레지스트에 있는 내용을 붙이게 된다.


vi
vi 는 visual edit의 준말이라는것 (어떻게 이것이 비쥬얼이란 말이 들어간다는 건지.. 그 이전 시대의 에디터를 설명하나 감이 오진 않는다). 올해 나이로 얼추 서른, 최장수 소프트웨어 중 하나란다. 이 책에서는 vim(Vi Improved) 를 사용하길 권장하고 이를 기준으로 설명한다.

- 명령모드 : 키 입력을 통해 vim에게 명령을 내리는 모드. 커서이동, 삭제, 복사, 붙이기 등 편집을 위한 모드.
- 입력모드 : 실제로 문서 입력 및 편집을 위한 모드. 타이핑하는데로 화면에 출력.
- ex모드 : 라인 에디터인 ex 에디터(vi 의 바탕이 된 소프트웨어)의 기능을 사용하는 모드. 파일 저장, 종료, 치환 등의 기능
명령 앞에 숫자를 넣게 되면 그 명령을 앞에 누른 숫자 만큼 반복한다는 의미가 있다. 예를 들어 10x 를 누르게 되면 x를 10번누른 효과, 즉 10개의 글자가 삭제되며, 10dd를 누르면 10개의 행이 삭제 된다. 또 다른 예를 보자면 10yw를 누른뒤에 2p를 누르게 되면 10개의 단어가 복사 되고 복사한 내용을 두번 붙여넣게 된다.
 붙여넣기를 이해하기 위해서는 레지스트를 알아야 한다. vi 는 총 17개의 레지스트를 갖고 있으며 이를 보기 위해서는 ex모드에서 :reg 명령을 넣으면 된다. 명령모드에서 "1p를 누르게 되면 1번 레지스트에 있는 내용을 붙이게 된다.
붙여넣기를 이해하기 위해서는 레지스트를 알아야 한다. vi 는 총 17개의 레지스트를 갖고 있으며 이를 보기 위해서는 ex모드에서 :reg 명령을 넣으면 된다. 명령모드에서 "1p를 누르게 되면 1번 레지스트에 있는 내용을 붙이게 된다.v를 누른 후 방향키나 h j k l 를 이용하여 커서를 이동하면 블록을 형성할수 있다(마우스 우클릭 효과). ctrl + v 를 누르면 사각형의 블록을 지정하게 된다. 이후 d 나 y를 누르면 삭제 복사가 이루어 지고 <는 탭 제거, >탭 삽입.
되돌리기는 u 되살리기는 ctrl + r.
치환은 ex 모드에서 :[범위]/[매칭문자열]/[치환 문자열]/[행 범위]. 예를 들어 :2,4s/old/new/g 2,4s는 2행에서 4행을 의미하며 g는 행 전체에 걸쳐 매치되는 문자열을 치환하라는 의미이며 :-1,+3s/old/new/ 에서 -1,+3은 현재 커서에서 위로 1행 아래로 3행을 뜻하고 마지막에 g가 없으므로 행에서 첫 번째 매치 문자열만 치환하라는 뜻이다.
-vi 의 고급 테크닉-
정규표현식:
vi 는 정규 표현식을 지원한다.
내가 정규식에 약하기 때문에 (항상 공부하겠다고 하고 스킵한 이유로...) 이 부분은 자세히 하고자 한다.

[a-z]\+[0-9]\+ 는 소문자 하나 이상 숫자 하나이상을 의미 하는 것으로 'like0101' 이나 'korea1117'을 의미 한다. OK\ [a-z]* 은 OK 문자뒤 하나의 공백 뒤에 소문자가 0개 이상 반복을 의미한다. 곧 'OK like'를 의미한다. ^[^0-9]\+.*[a-z]$ 에서 괄호 앞의 ^ 행의 시작을 의미하며 괄호 안의 ^는 not을 의미 하고 \+는 1 이상, .은 '\n'을 제외한 문자를 *는 0개 이상, $는 행의 끝을 의미하므로 숫자로 시작하지 않는 행이며 행 가운데는 아무 문자가 있을수도 있고 끝이 소문자로 끝나는 행 을 지징하게 된다. \
{kind=link}

vi는 POSIX 문자 클래스를 지원한다. 또한 확장 정규 표현식도 지원한다. 이는 생략하기로 한다.
여러 파일 편집시 예를 들어 vi를 이용해 확장자가 .py로 끝나는 파일을 열었다고 했을때(vi *.py) 열린 파일 리스트를 보기 위해서는 :ls 는 이용해 가능하고 지정 파일로의 이동은 :b2 (두번째 파일 버퍼를 의미한다) 식의 ex모드 명령어로 가능하다. :map 명령어는 단축키 지정시 사용한다.
vim 설정하기 : 사용자 홈 디렉토리에 .vimrc 파일을 생성하게 되면 vim은 실행 할때 이 파일을 읽어서 수행하게 된다. 그래서 .vimrc 파일을 이용해 원하는 사용자에 맞도록 vim를 설정할 수 있다. .vimrc파일에서 주석은 "로 시작한다.
매크로의 사용 : 예를 들어 perl로 코딩을 했는데 python에 익숙한 나머지 행 마지막에 ; 를 빼먹었다. 그러면 커서를 문장의 맨뒤로 보낸뒤 입력모드로 바꾸고 ;를 입력한다음 행을 바뀌야 한다. 이를 수행하는 과정을 나열하자면 $ i ; Esc Enter 이다 (물론 $ i 다음에 그냥 행아래로 내려가서 계속 ;를 넣으면 가능한긴하다). 이를 매번 치기 귀찮으니 명령어 과정을 vim에서 제공하는 레지스트(a-z 레지스트) 에 저장해서 이용할수 단축키로 이용 할수 있다. 명령모드에서 qb를 누르면 b레지스트에 기록시작을 뜻하며 $ i ; Esc Enter 를 순서대로 입력수 q를 누르면 b레지스트에 저장이 된다. 그뒤 다시 명령모드에서 5@b 라고 치면 b레지스트에 있는 매크로가 5번 수행하는 결과를 가져와서, 곧 5개의 행 뒤에 ;를 붙인다.
다중창 사용하기 :
Ctrl+w n : 가로 분할로 새창이 띄어짐, 여기서 :e 명령어로 파일을 불러올수 있다.
Ctrl+ww : 창간커서 전환
Ctrl+w s : 현재 파일을 가로로 분할, 같은 파일의 다른 부분을 참조할 때 이용한다.
Ctrl+w v : 현재 파일을 세로로 분할
-vim 의 조력자들 ctags, cscope, 기타 플러그 인-
연동 프로그램은 바이너리 형식의 실행 파일로 vim과 독자적으로 동작할수 있다. ctags, cscope, man등이 있다.
내장 스크립트는 vim이 기능확장을 위해 자체적으로 가지고 있는 것으로 vim 플러그 인아리 부르고 .vim의 확장자를 갖는 것으로 bufexplorer, taglist, calendar, vimspell등이 있다.
ctags :
-알아두면 유용한 팁-
파일 탐색 : :r !ls 는 원래 :!를 이용해 쉘명령어를 이용할수 있는데 :!ls를 하면 vim상태에서 ls를 실행하게 된다. 결국 ls 명령어로 출력되는 결과를 현재 커서의 위치에 삽입하라는 명령어가 된다.
:20vs ./ 이는 explorer 플러그 인이 설치 되어 있어야 한다. 파일의 리스트를 보고 싶을때 !ls를 이용할 수도 있지만 :20vs 를 이용하여 선택하면 바로 열리게 된다. 뒤의 ./는 현재 디렉토리를 뜻한다.
폴딩 기능 : 보기 싫은 긴 함수 같은 경우 '{' 위에 커서를 두고 v]}zf 명령어를 치면 함수가 폴딩되고 zo를 치면 다시 풀리게 된다. 사실 좀더 일반적으로 v로 영역을 지정하여 zf를 누르면 이부분이 폴딩된다. v는 앞에서 배운 드레그의 의미이다.
여러 행 앞에 탭 끼워넣기 : [N]>> 는 커서 위치서부터 N 행에 탭넣기 [N]<< 탭제거. 예를 들어 7>> 이면 7행 앞부분에 탭넣기가 된다.
자동 완성 기능 : 코딩중에 앞에서 사용한 함수 이름이나 변수를 일부만 치고 Ctrl+p를 누르면 앞에서 사용된 변수들 중 입력한 문자가 들어가는 변수들이 리스트업 된다.
Subscribe to:
Comments (Atom)